
About This Database
We aimed at grasping the diversity of microorganisms and their changes
in the coastal areas of Tohoku sea, where the huge earthquake and tsunami hit.
They developed a method to monitor relationships between microbial flora and the
environment by comprehensive analysis of metagenomic DNA.
In this database, time series data sampled from Sendai Bay (a regional coast in
Tohoku area), A-line (a offshore in Tohoku area) etc from 2012 to 2016 are
stored.
In addition to the nucleotide sequences, related information such as environmental
information, analysis methods etc.
are included in the database.
Furthermore, microbial community information of each sample calculated from the
sequencing analysis can also be browsed as graphical images.
List of data
- Physical and biological information at each sampling spot for comparison of environment
- Nucleotide sequence data (Sendai Bay, A-line, Ofunato Bay)
- Interactive time series graphs
- Blast search to show the avandance of assembled contig sequences
- Methods (DNA extraction, NGS sequencing, etc)
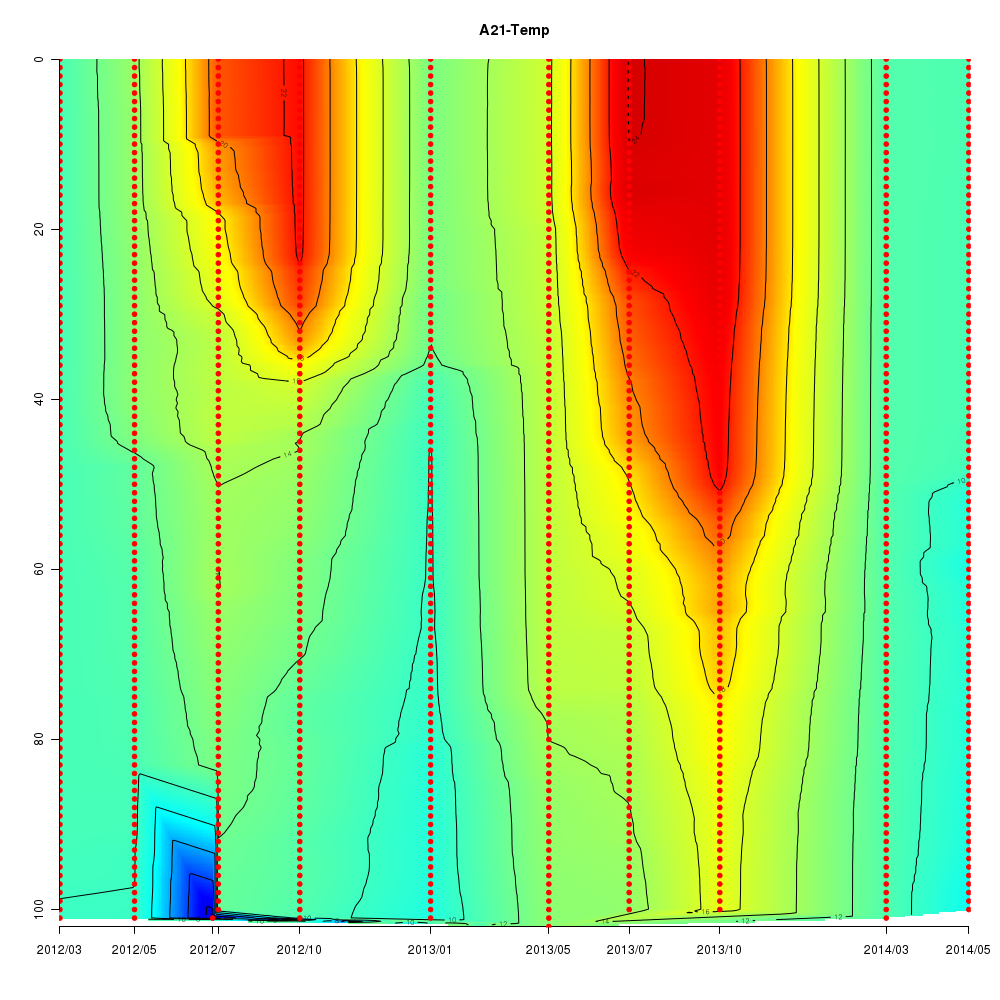
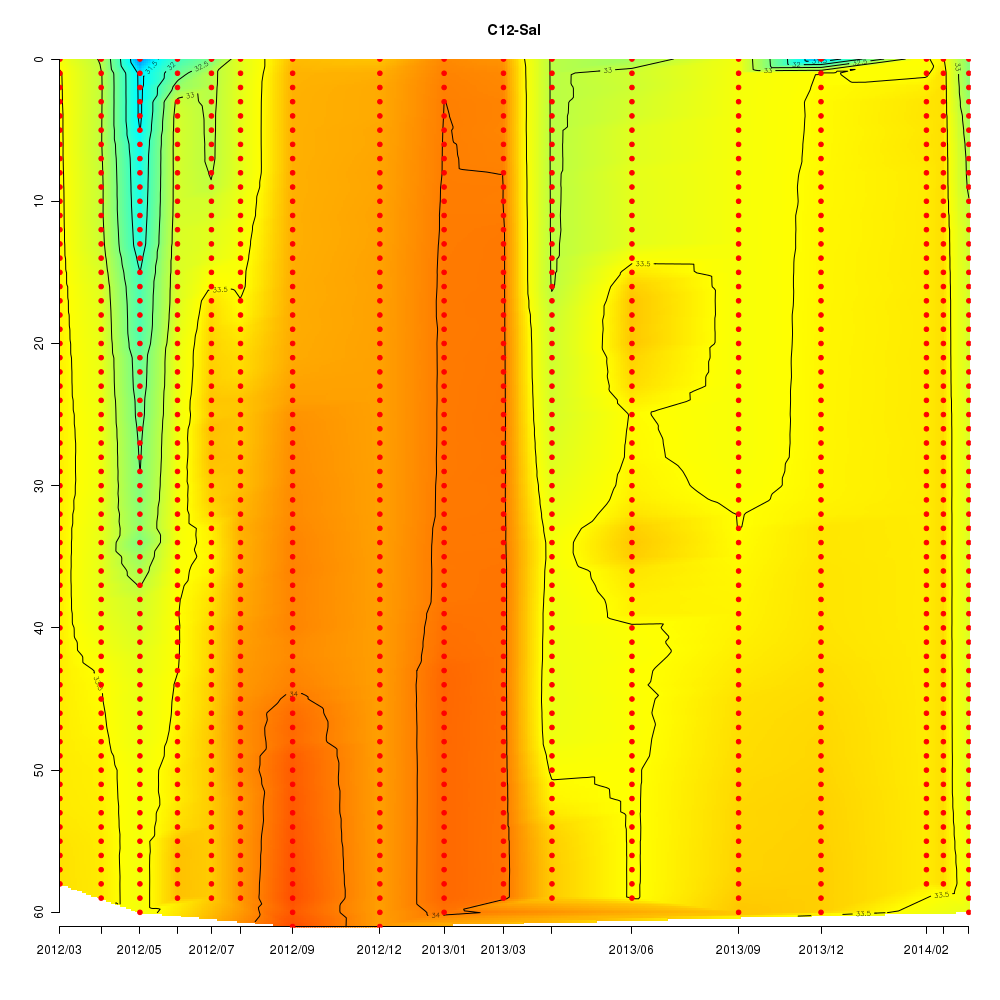
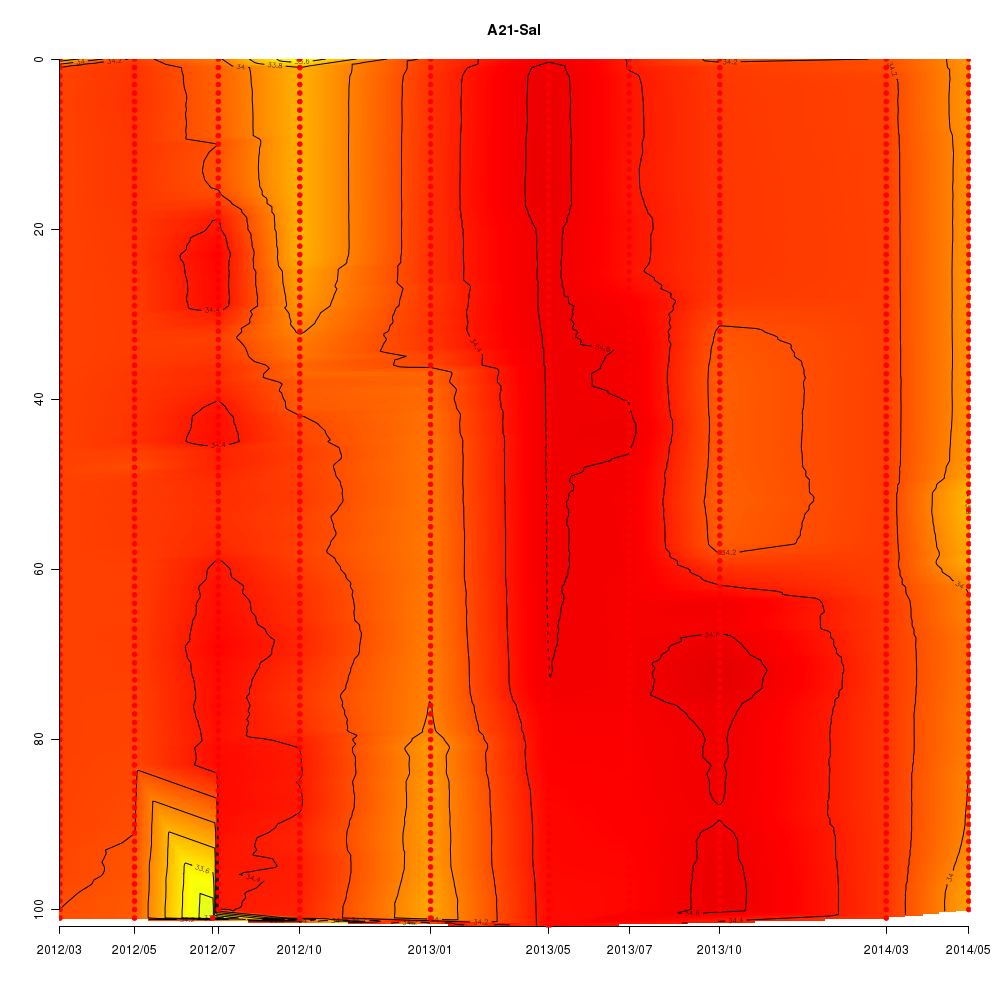
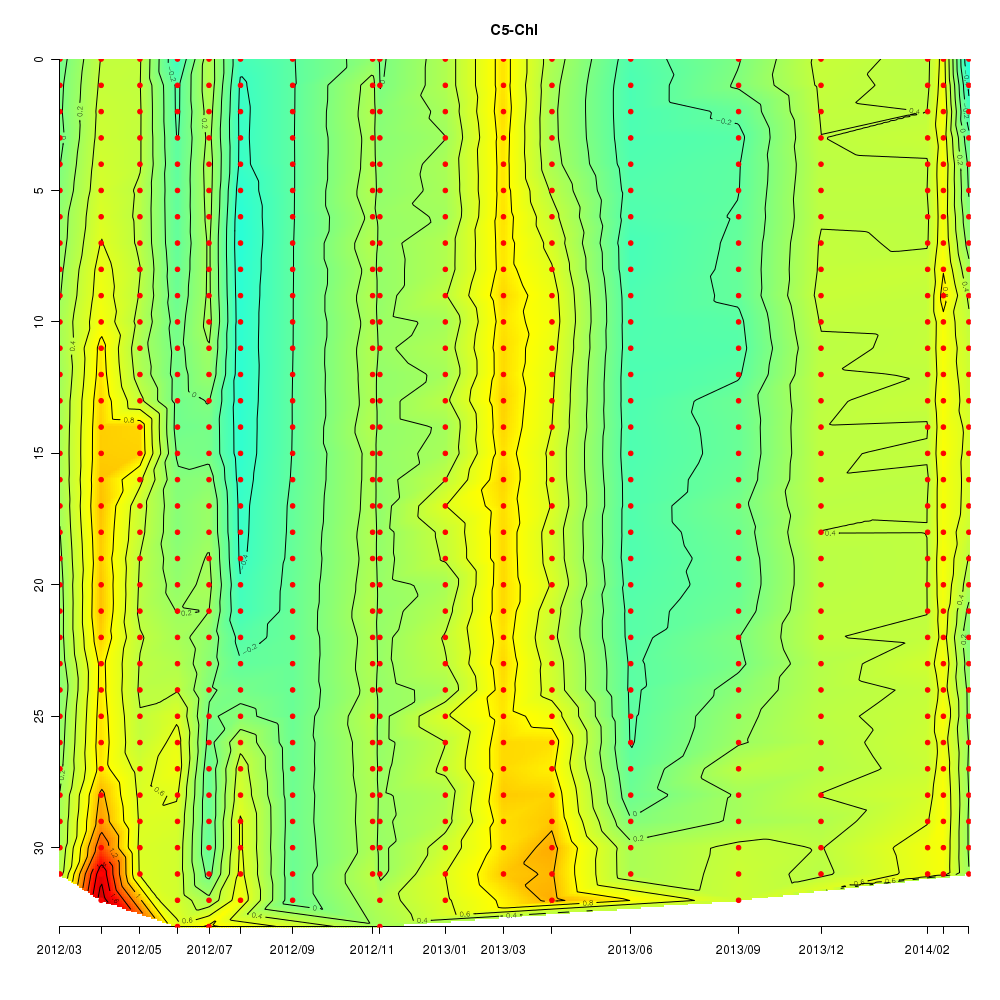
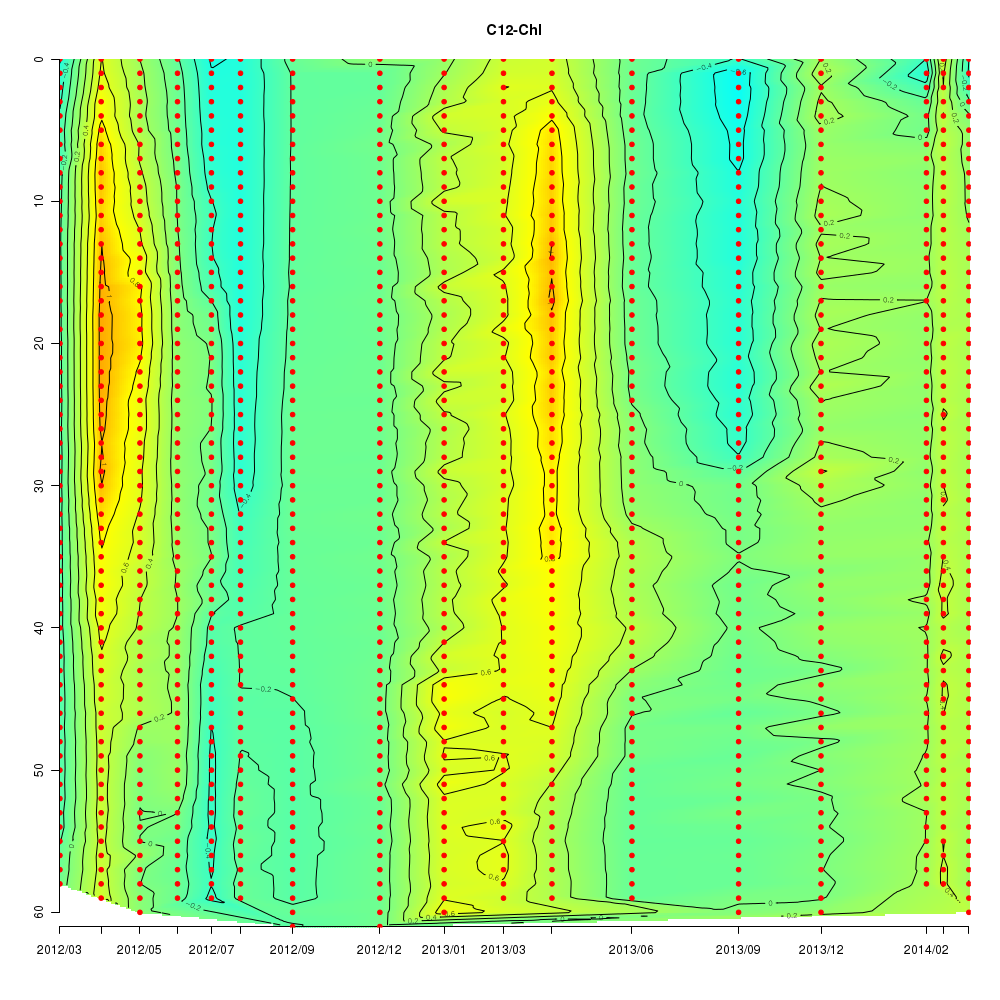
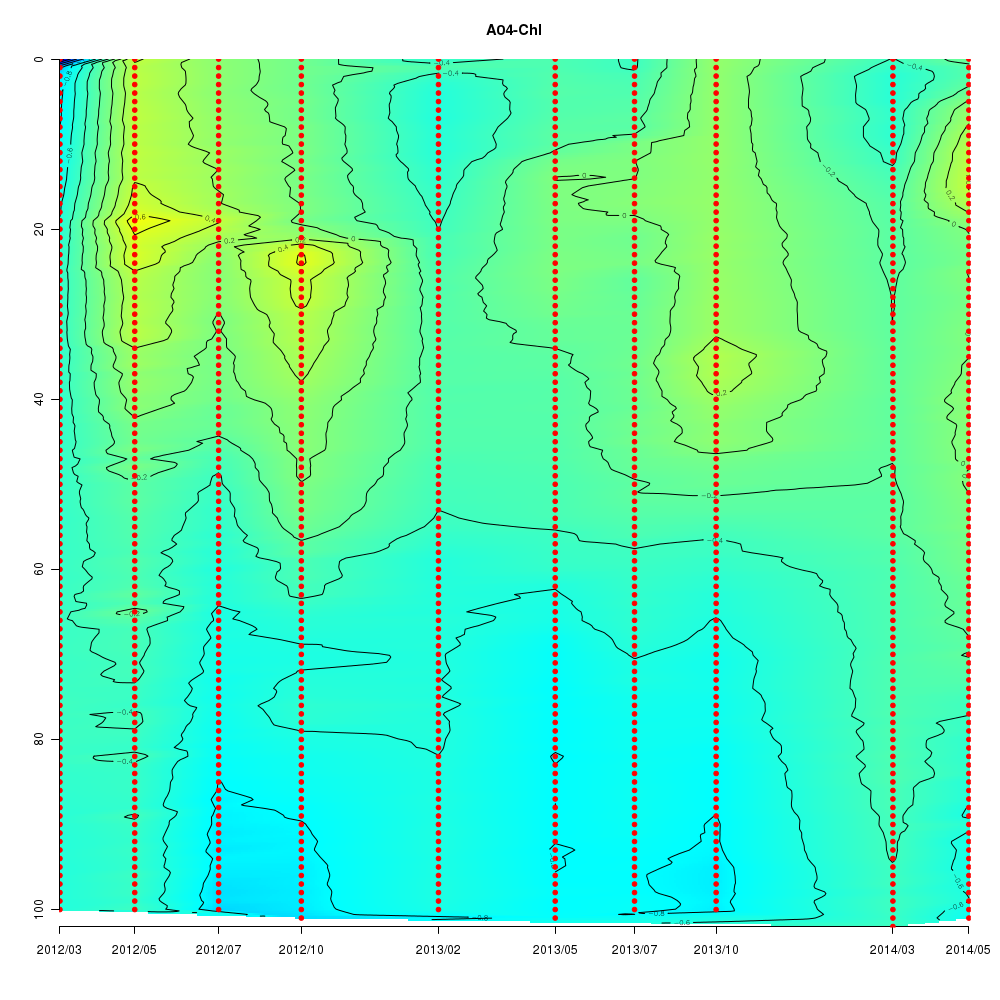
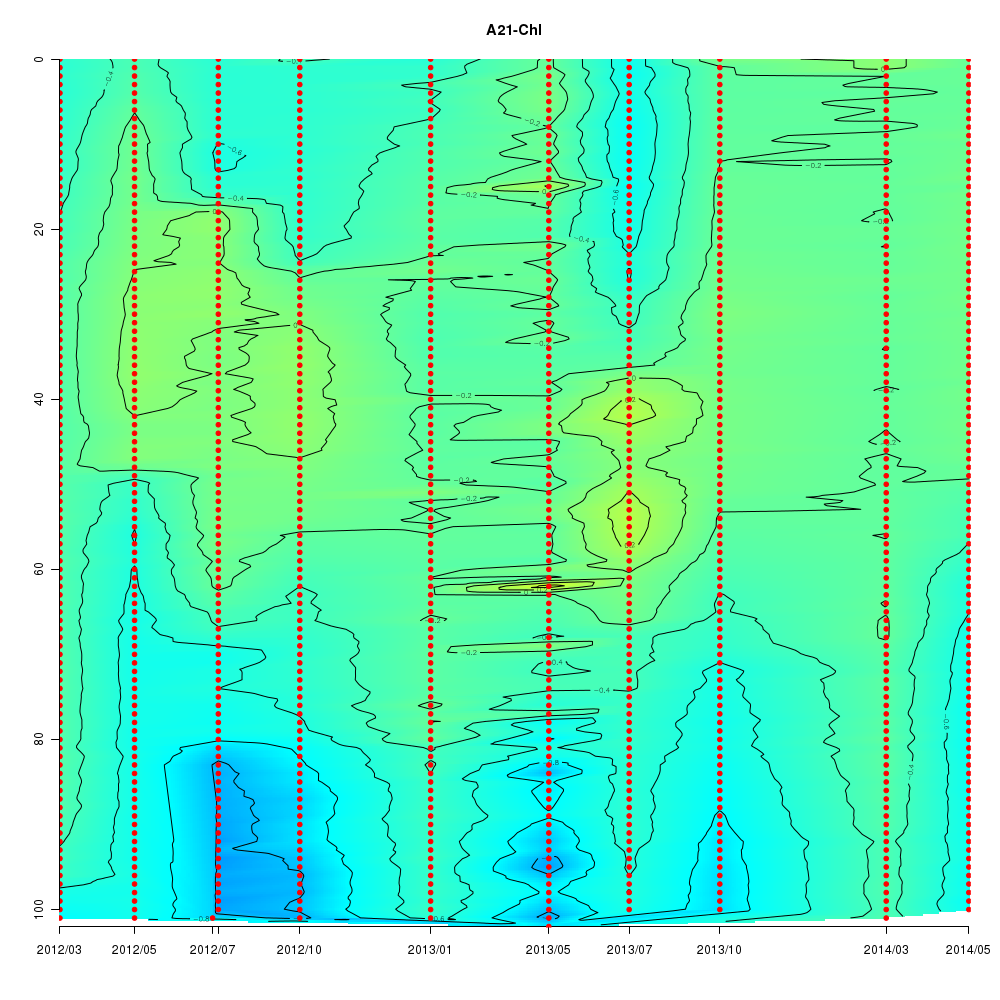
Environmental information
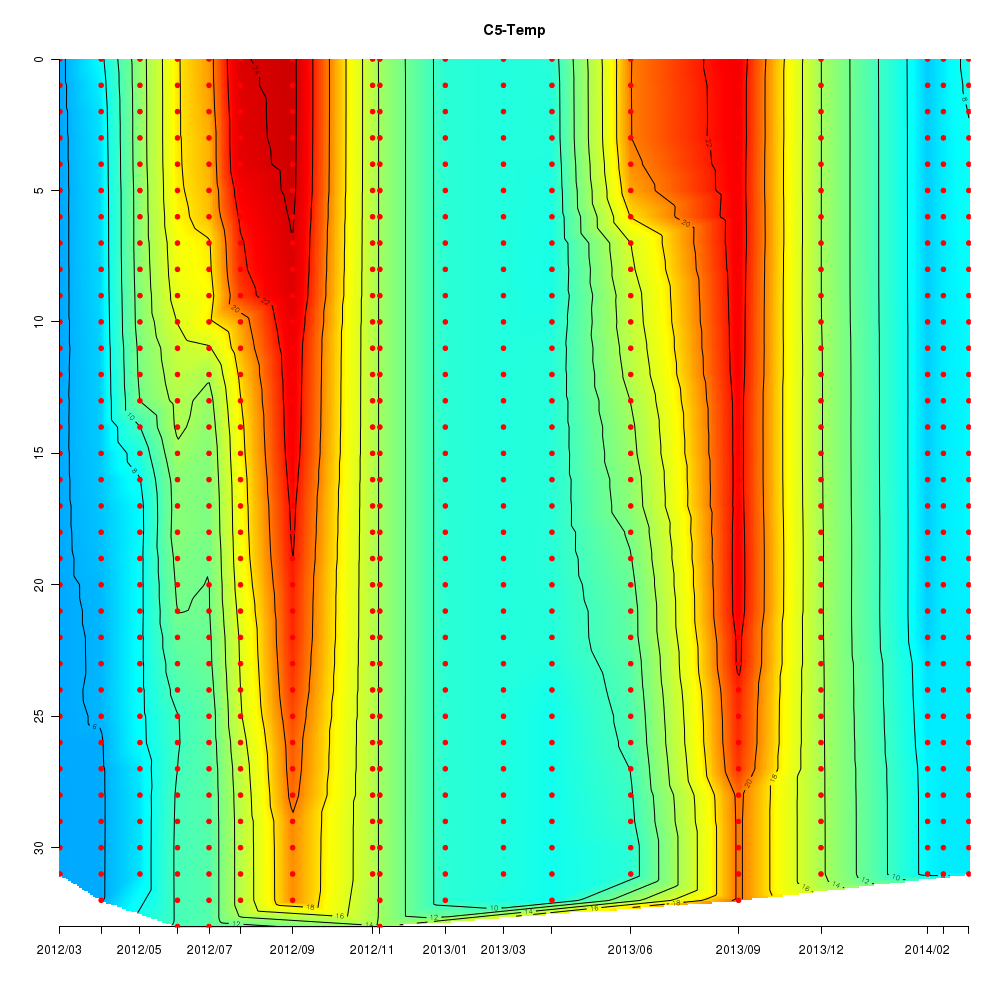
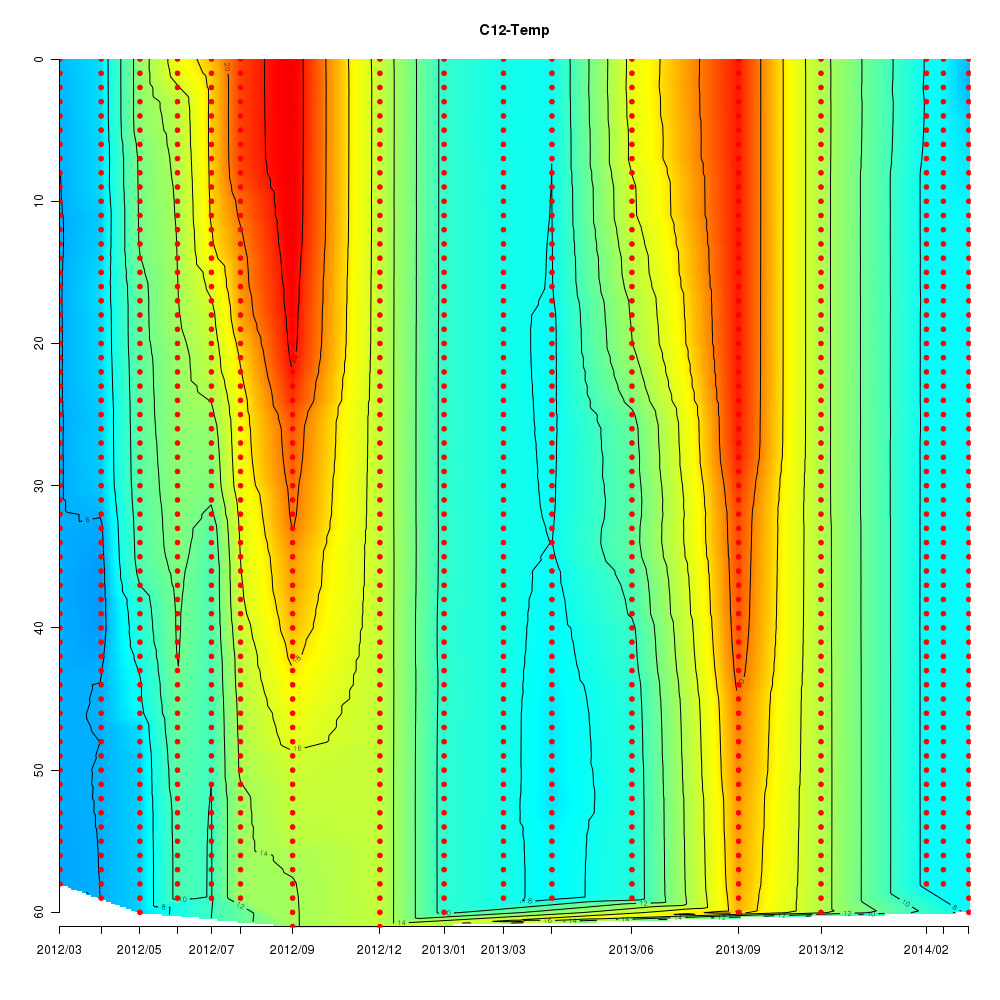
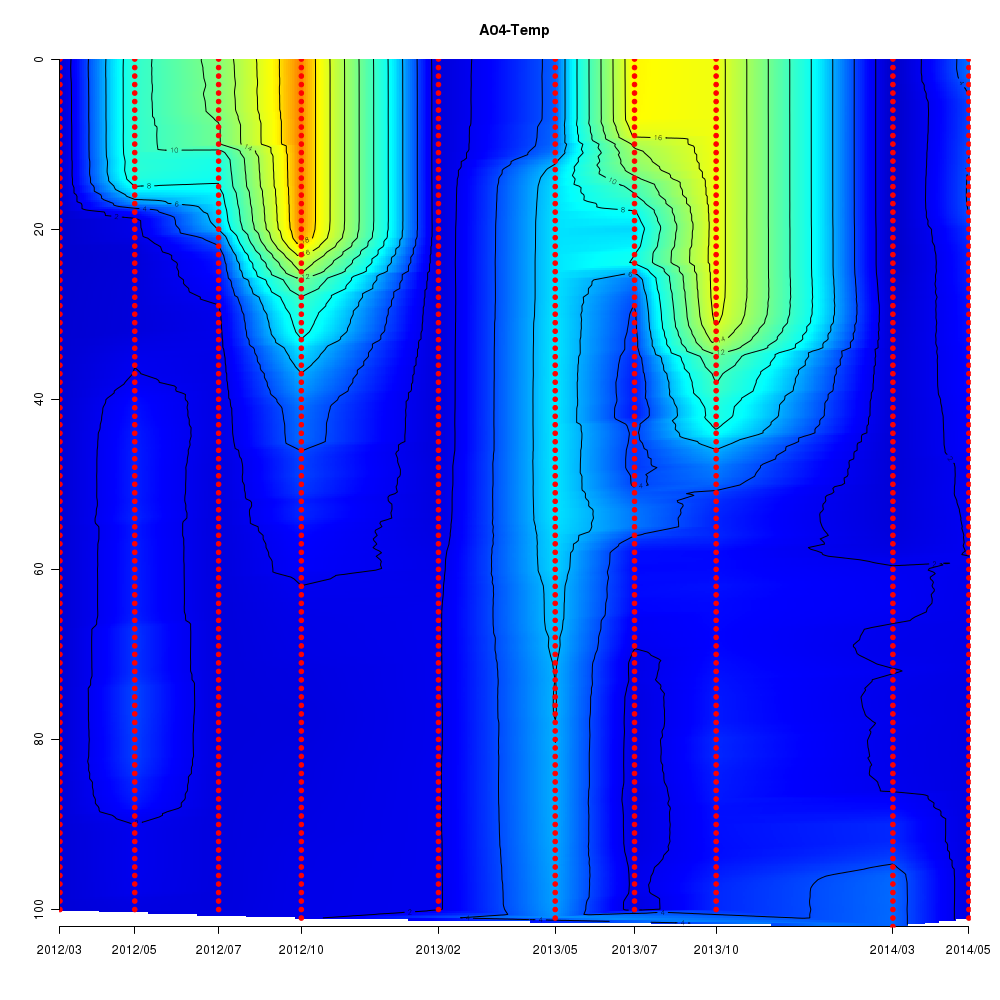
| Sendai-Bay C5 | Sendai-Bay C12 | Aline A4 | Aline A21 | ||
|---|---|---|---|---|---|
| Temp. (℃) |
|
 |
 |
 |
|
| Sal. | 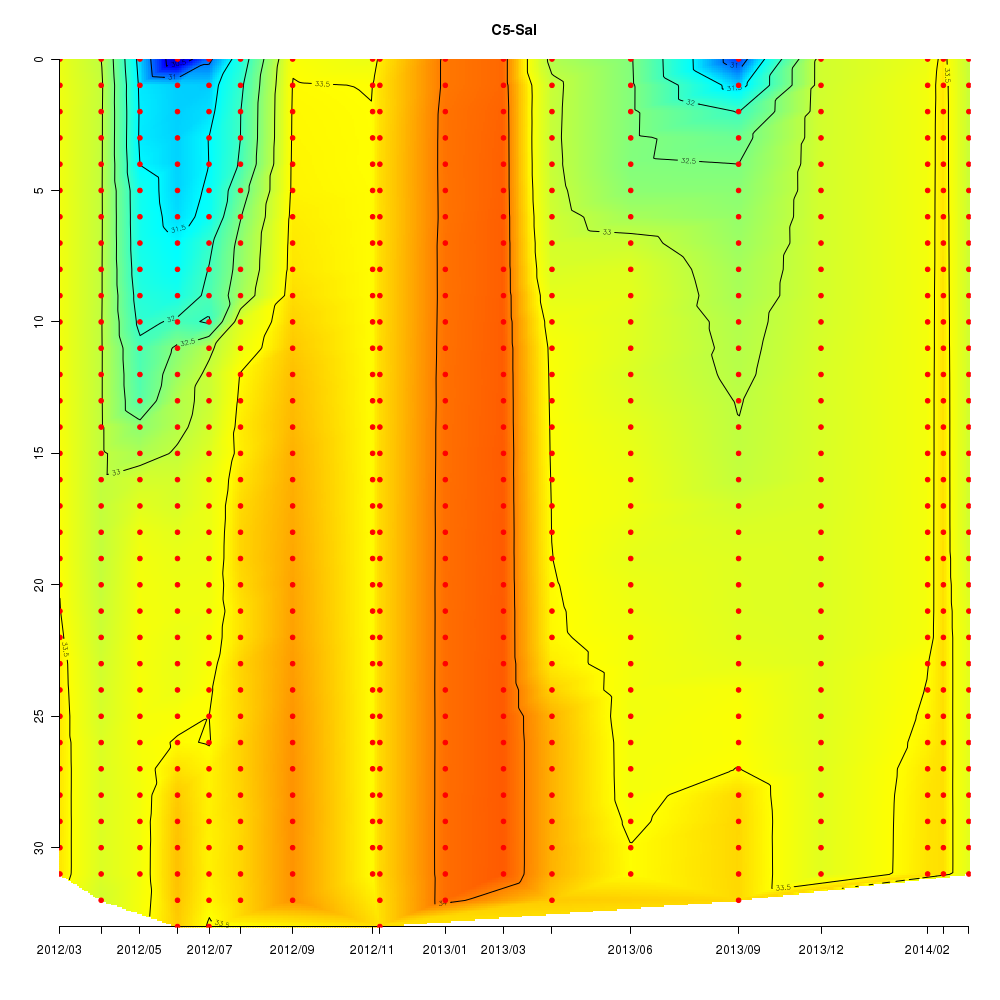
|
|

|
|
|
| Chl. (µg/l) |
|
|
|
|
|
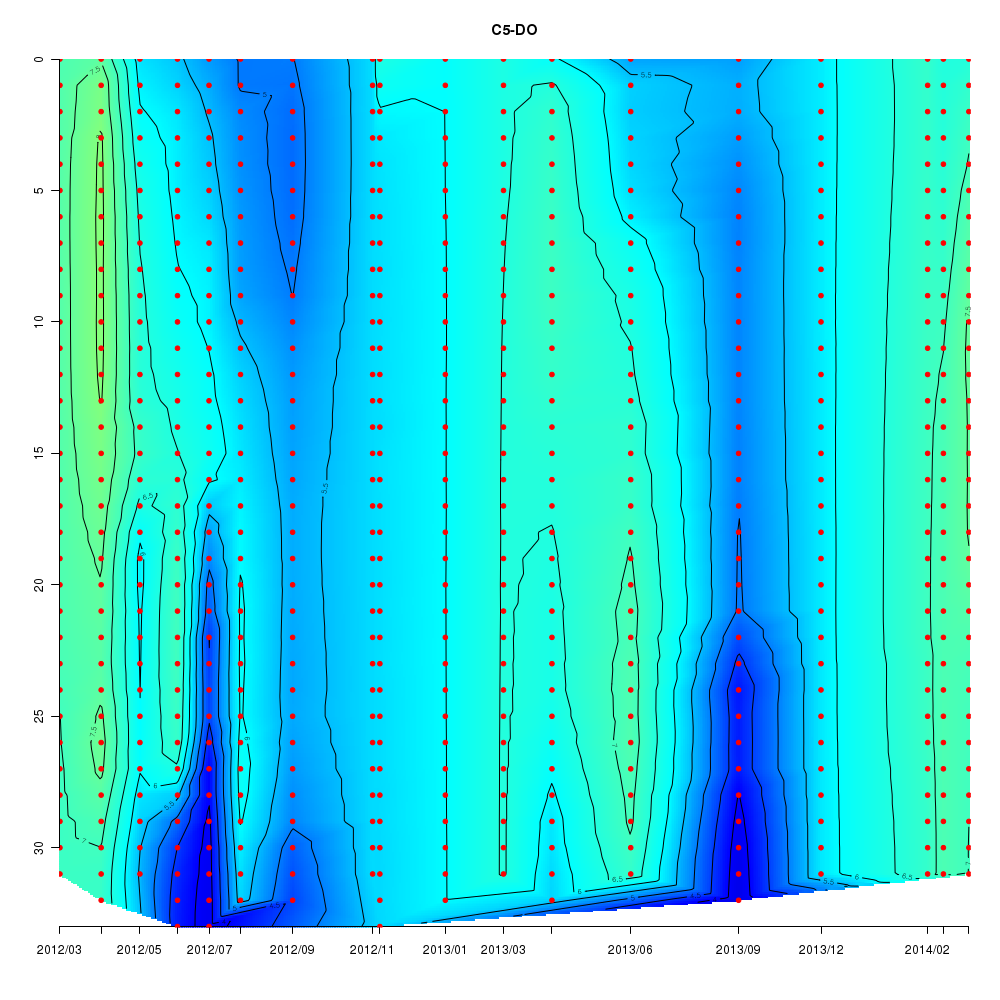
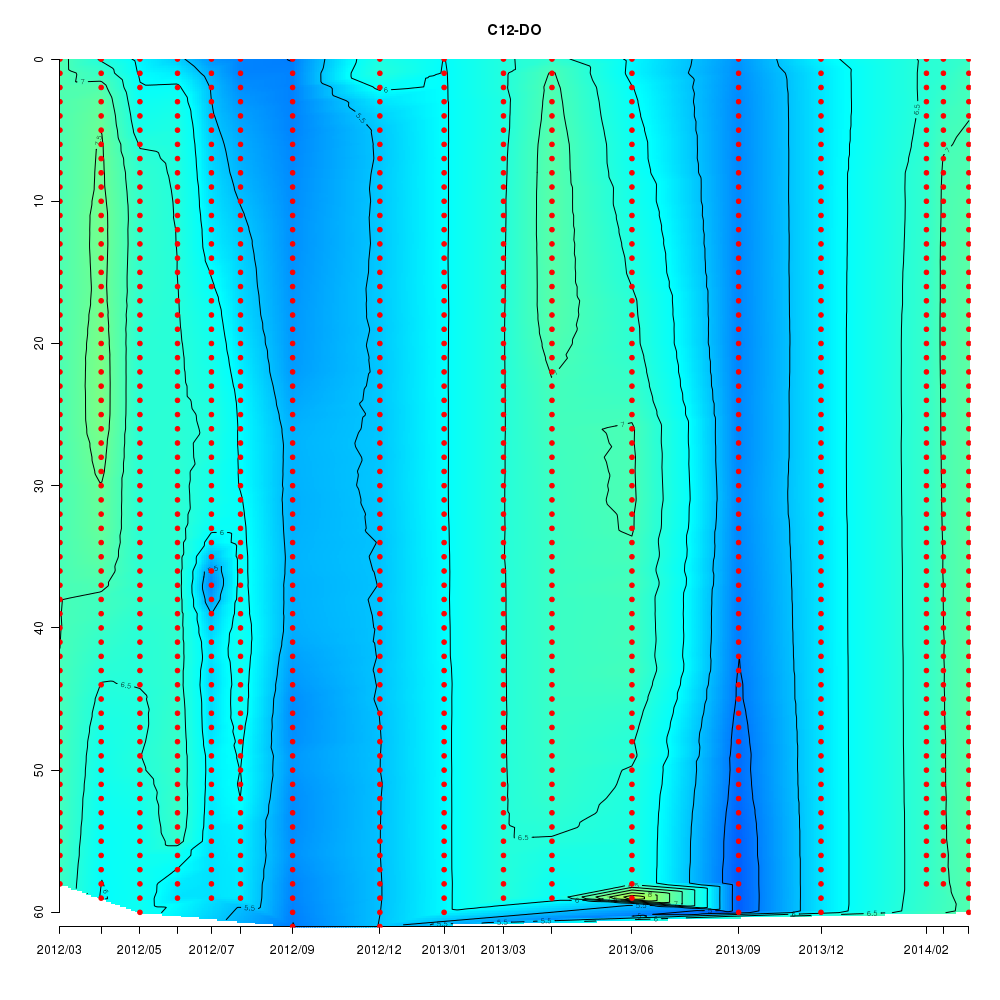
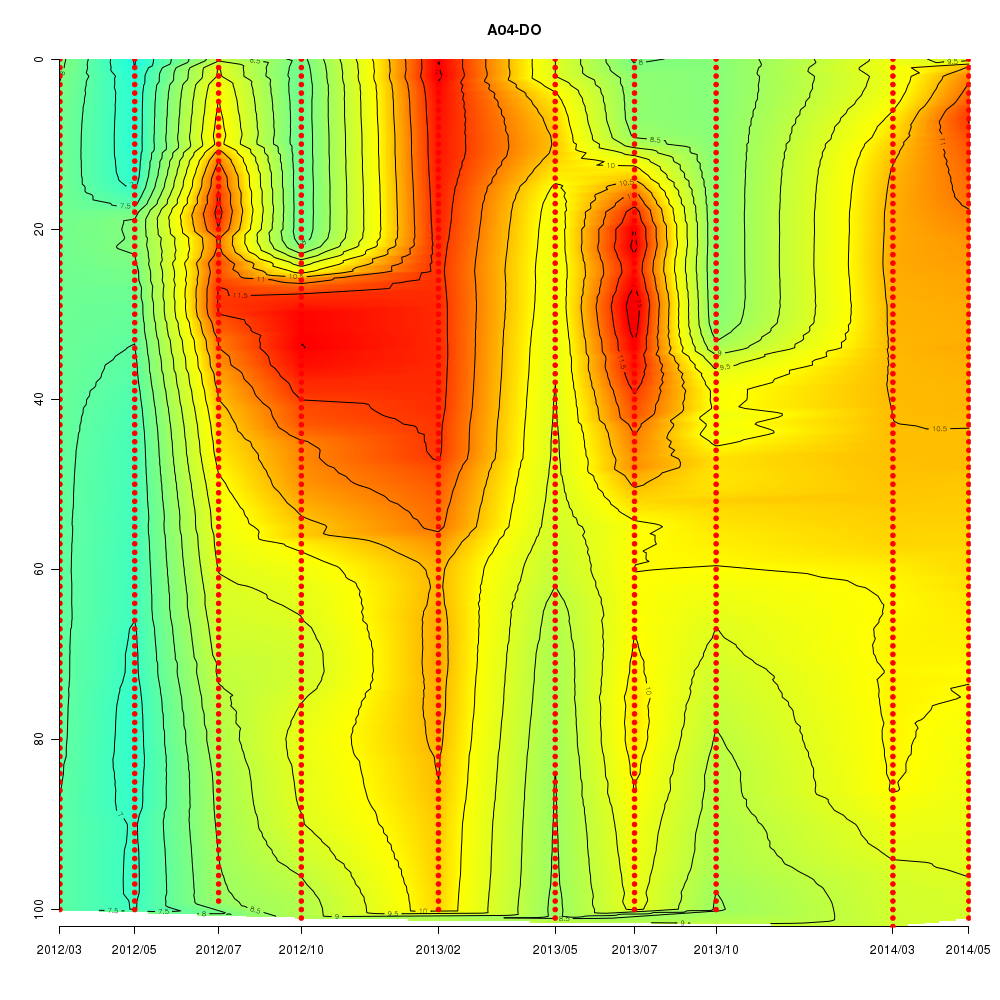
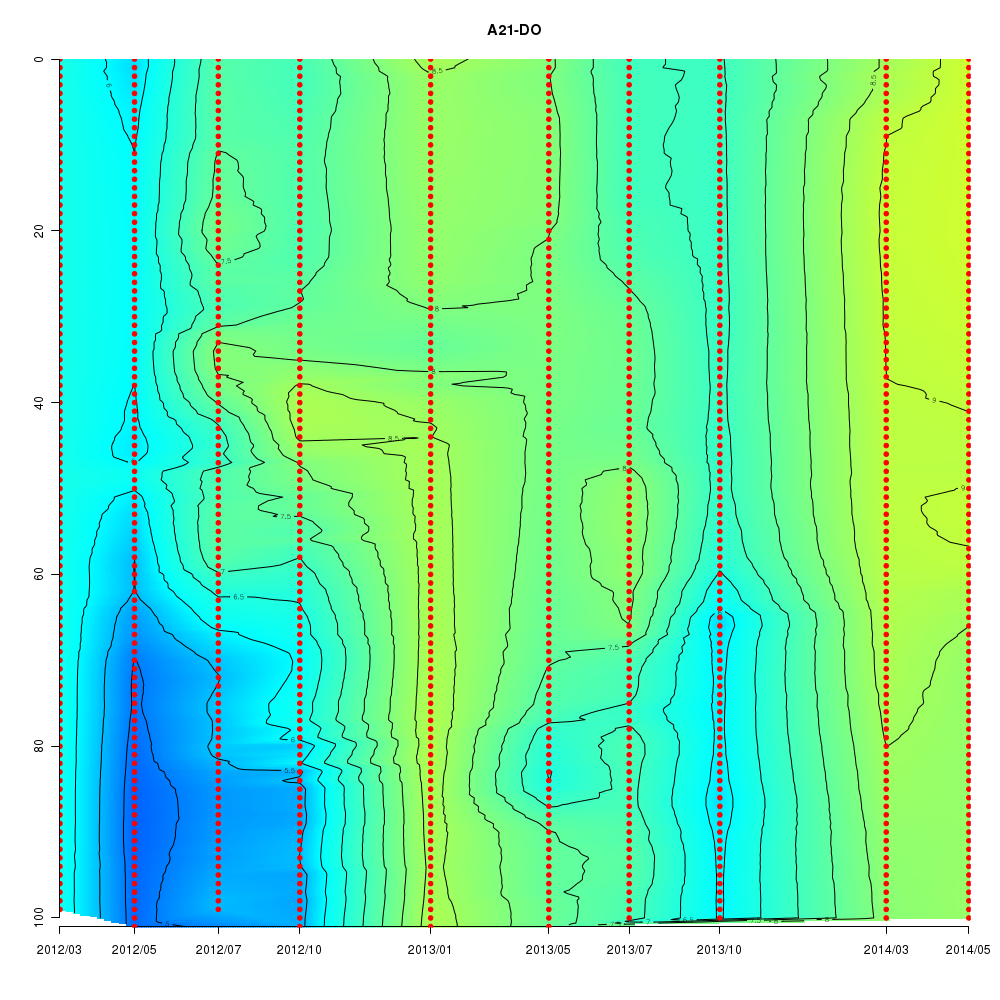
| DO (µmol/kg) |
 |
|
|
|
(x: date, y: depth)
Data
Blast search
Raw data
Interactive time series graphs
- Time series of SILVA classification using 16S rRNA in the shotgun data
- Time series of NCBI NT classification of the shotgun data
- 3D time series of bacteria by 16S amplicon using SILVA database (2012 - 2014 Sendai-Bay C5)
- 3D distribution of bacteria in Sendai-Bay 2014/04/16 by 16S amplicon using SILVA database
Materials and Methods
Shotgun data
Sampling- 100 µm filter passed and 20 µm filter captured microbiome
- 20 µm filter passed and 5 µm filter captured microbiome
- 5 µm filter passed and 0.8 µm filter captured microbiome
- 0.8 µm filter passed and 0.2 µm filter captured microbiome
Analysis
Sequenced reads were trimmed with a minimum quality value of 15 by FASTX_Toolkit. Trimmed reads were aligned to NCBI NT database or SILVA database (SSU_Ref_NR ver. 119) by blastn with default e-value and a minimum bit-score value of 100, and its taxonomy was identified by the lowest common ancestor (LCA) method using in-house script.
Bacteria amplicon 16S data
Sample collection and DNA extractionWater samples were cooled immediately after collection and kept in dark. For the over 0.2 μm fraction samples, waters of 300 to 500 mL were filtered through a membrane filter of normal pore size 0.22 μm (GS, Millipore) within 10 h after sample collection. Filters were stored frozen at −80 °C until processing. To avoid an inhibitive effect of high concentration of suspended matters in the bottom water, DNA was extracted using a FAST DNA-SPIN kit for soil (MP. Biomedicals) in accordance with the manufacturer's instructions. For the 0.2 - 0.8 μm fraction, the same DNA with the shotgun analysis were used.
Multitag pyrosequence analysis
First PCR was conducted using 27Fmod and 338R primers (Kim et al., 2013) modified by adding 33 and 34 bp nucleotide respectively to add molecular identifier (MID) tags later, with an initial denaturation at 95 °C for 2 min, followed by 20 cycles of denaturation at 95 °C for 30 s, annealing at 52 °C for 30 s, and extension at 72 °C for 30 s; final elongation was carried out for 1 min at 72 °C. Product was visualized on an agarose gel with SYBR® Safe (Life technologies), excised and purified from the gel and quantified by using a QuantiFluorTM (Promega). Approximately 10 ng of the first PCR product was used as template for the second PCR conducted to add MID tags using a forward (59 bp) and reverse (54 bp) primer set with an initial denaturation at 98 °C for 30 s, followed by 12 cycles of denaturation at 98 °C for 10 s, annealing at 60 °C for 30 s, and extension at 72 °C for 30 s; final elongation was carried out for 1 min at 72 °C. The PCR programs were determined in accordance with the sequencing machine operating company's instruction (FASMAC Co. Ltd). Sequencing was performed on the Illumina MiSeq by 300 bp paired end run.
Data Analysis
Homology search with blastn was performed on SILVA database, blastn outputs with bit score larger than 100 were extracted, and microbial flora was identified by LCA method.
References
- Kim, S.W., Suda, W., Kim, K., Oshima, K., Fukuda, S., Ohno, H., Morita, H., Hattori, M., 2013. Robustness of gut microbiota of healthy adults in response to probiotic intervention revealed by high-throughput pyrosequencing. DNA Res. 20, 241–253.
- Sakami, T., Watanabe,T., Kakehi, S., Taniudchi, Y., Kuwata, A., 2015. Spatial variation of bacterial community composition at the expiry of spring phytoplankton bloom in Sendai Bay, Japan. Gene 576, 610-617.
Pico- and nanophytoplankton amplicon-data (18S rDNA)
SamplingSurface seawater was collected by Niskin bottle and pre-filtered through a 5 µm-mesh- size plankton net to remove large plankton. Pico- and nanophytoplankton cells in the filtrate were concentrated to 40–100× by tangential flow filtration equipped with a regenerated cellulose membrane (100kD MWCO). The concentrated cells were gently frozen from room temperature to –35ºC in a cryoprotectant (10% Dimethyl sulfoxide), and cryopreserved at –196ºC in liquid nitrogen (2-step freezing method; Morris 1978) ("Frozen" treatment). For the other treatment of "non-Frozen", this cryopreservation step was skipped. The phytoplankton cells were detected by comparing the side angle scatter of the chlorophyll a fluorescence-positive cells with that of fluorescent polystyrene beads (3-µm diameter), and 2500 of them were sorted per sample using a flow cytometer equipped cell sorter (EPICS ALTRA, Beckman Coulter).
Library preparation and 454 sequencing
Genomic DNA was extracted using an alkaline lysis method (Zhang et al. 1992), and amplified with Genomiphi V2 in order to obtain sufficient amount of DNA for downstream procedure. Eukaryotic 18S rDNA was PCR amplified using the primer set of 545F and 1119R (Kawachi et al. 2016) attached to multiplex identifier (MID). The amplicons of ca. 580 bp were separated in agarose gel and purified using QIAquick Gel Extraction Kit (Qiagen), and analyzed by GS Jr. 454-pyrosequencer.
OTU clustering and classification with SILVA by LCA
Forward-oriented reads were separated according to the MIDs, quality controlled using a minimum quality value of 27, and cleaned by removing potential chimera sequences that would be generated during PCR. Next, the operational taxonomic units (OTUs) were established using a 95% sequence similarity level by assembler (Assams-assembler in claimant, Tanabe et al. 2013). The representative sequence in each OTU was aligned to SILVA alignment (SSU_Ref_NR ver. 123), and its taxonomy was identified by the lowest common ancestor (LCA) method using a SINA program (Pruesse et al. 2012).
References
- Kawachi, M., Kataoka, T., Sato, M., Noël, M.-H., Kuwata, A., Demura, M., Yamaguchi, H., 2016. Application of cryopreservation to genetic analyses of a photosynthetic picoeukaryote community. Gene 576, 708–716.
- Morris, G.J., 1978. Cryopreservation of 250 strains of Chlorococcales by method of 2-step cooling. British Phycological Journal 13, 15-24.
- Pruesse, E., Peplies, J., Glockner, F.O., 2012. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823-1829.
- Tanabe, A.S., Toju, H., 2013. Two new computational methods for universal DNA barcoding: A benchmark using barcode sequences of bacteria, archaea, animals, fungi, and land plants. PLoS ONE 8, e76910.
- Zhang, L., Cui, X.F., Schmitt, K., Hubert, R., Navidi, W., Arnheim, N., 1992. Whole genome amplification from a single cell - implications for genetic- analysis. Proc. Natl. Acad. Sci. USA 89, 5847-5851.
Enumeration of phytoplankton
SamplingFor phytoplankton analysis, 1-L seawater samples were immediately fixed with acid Lugol’s solution (4 % final concentration) and preserved at 4 °C. Samples were con-centrated by reverse filtration through 2-μm nucleopore filters.
Analysis
In the concentrated samples, diatom species were identified following Hasle and Syvertsen (1996) and their abundance (cells per liter) was estimated using light microscopy.
References
- Hasle GR, Syvertsen EE (1996) Marine diatoms. In: Tomas CR (ed) Identifying marine diatoms and dinoflagellates. Academic, San Diego, pp 5–385